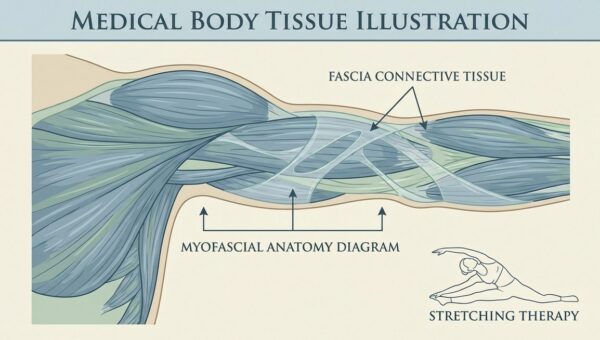
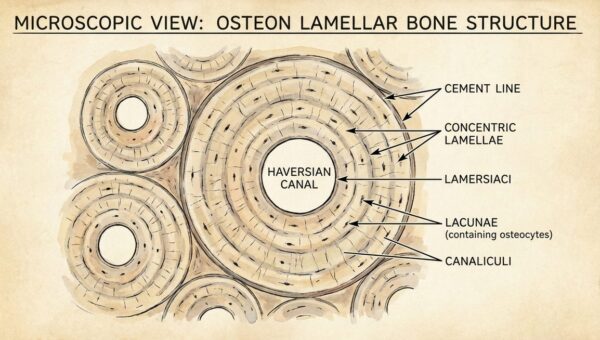
Die Schwesternhaube — Geschichte, Symbolik und Wandel eines Berufssymbols der Pflege
Die Schwesternhaube — Geschichte eines Berufssymbols Die weisse Haube auf dem Kopf einer Krankenschwester ist eines der …
Weiterlesen